
I denne artikel finder du ikke svar på, om din software skal have en CE-mærkning. Du får til gengæld en lynhurtig indflyvning til nogle af de væsentligste regulatoriske krav og gode råd til, hvad du med fordel kan gøre, når du skal undersøge om din software skal CE-mærkes.

Skal CE-mærkning tages seriøst?
CE-mærkning af din software er en vigtig markør for kvaliteten af den software, du sender på markedet, og er indført som en sikring for borgeren, slutbrugeren og patienten. Sundhedsfaglige beslutninger er i stigende grad afhængige af software, og for at værne om patientsikkerheden er der derfor ekstra opmærksomhed på kvalitet, risiko og sikkerhed. Med EU forordningen (EU) 2017/745 (MDR) er kravene for CE-mærkning af medicinsk udstyr – herunder software med medicinsk formål – skærpet betydeligt. I praksis betyder det bl.a., at en meget større del af det, som kaldes medicinsk software, skal certificeres.
Hvad er medicinsk software?
Hvis din software har et medicinsk formål, enten i sig selv eller som en del af et medicodevice, så skal den CE-mærkes. Om din software kan karakteriseres som medicinsk udstyr afhænger af “intended use” – altså dit tiltænkte formål. Fx er det tiltænkte formål med den træpind, som din læge anvender til svælgundersøgelse det, der kvalificerer den som medicinsk udstyr – selvom den til forveksling ligner en ispind.

Helt så simpelt er det dog ikke. Når du har fundet ud af om din software er medicinsk software, skal det klassificeres indenfor én af de fire risikoklasser. Klassificeringen afspejler den risiko, der er forbundet med at anvende produktet; jo større risiko, jo højere risikoklasse. Derfor vil et stykke software, som leverer beslutningsstøtte til den kliniske behandling medføre, at softwaren kommer i en høj risikoklasse, hvor den skal certificeres af et bemyndiget organ.
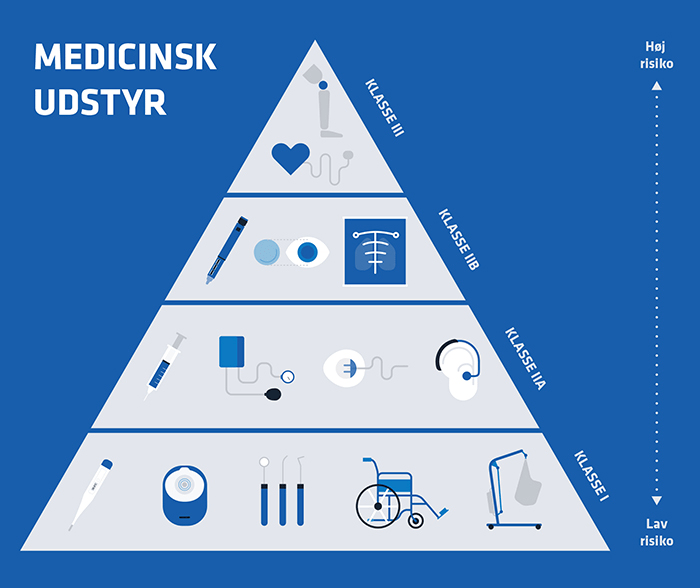
Hvor meget haster det?
Processen til CE-mærkning handler ikke blot om den sidste blåstempling af produktet med et CE-mærke. Som softwareleverandør er det helt essentielt at have styr på hele softwarens lifecycle i processen fra analyse, design, udvikling, test gennem fx anvendelse af system til kvalitetsstyring og være på forkant med den tekniske dokumentation. For alle produkter over klasse 1 handler det også om at være tidligt ude og alliere sig med et bemyndiget organ. At få fat i et bemyndiget organ kan nemlig være en udfordring i sig selv, da der er et fåtal af bemyndigede organer i hele EU. Så hvis du ikke vil bagerst i køen, skal du igang i dag.
Sådan kommer du videre
- Få overblik. Få styr på rammerne for CE-mærkning af medicinsk udstyr – start fx. ved Lægemiddelstyrelsen (tilsynsmyndighed i DK).
- Er du en ispind eller en tungespatel? Find ud af om din software kvalificeres som medicinsk udstyr – tjek EU-forordningen EU) 2017/745 (MDR) og kig særligt ind i klassificeringsreglerne bilag VIII, hvor software har fået sin egen “regel 11”.
- Skær ind til benet. Hvad er “intended use” / dit tiltænkte formål med produktet?
- Find din risikoklasse. Hvor placerer din software sig? Her kan du med fordel anvende EU’s vejledning, som indeholder beslutningstræ og eksempler på, hvordan software klassificeres.
- Start tidligt. Tænk kvalitetsstyring og risikostyring ind fra designfasen og vær på forkant med teknisk dokumentation og dokumentation af processen omkring udvikling og test.
- Hent hjælp. Orienter dig i ISO-standarderne. Det er ikke et krav i MDR at implementere ISO 13485 for medicinsk udstyr, men det er et rigtig godt værktøj til at sikre, at du lever op til kravene omkring kvalitetssikring i forordningen. På samme måde er ISO 14971 med til at holde dig på sporet i forhold til kravene for risikostyring.
- Ring til en ven. Kontakt med fordel nogen, som selv har været igennem processen for overensstemmelsesvurdering eller overvej et møde med Lægemiddelstyrelsen, som tilbyder 1-1 møde vedrørende regulatorisk vejledning.